Scientific and medical rationale
The primary cilium is a microtubule-based organelle protruding from the cell surface of most cell types to the extracellular medium. Its function is to sense environmental signals (mechanical strain or molecules) and control key signaling pathways during development and tissue homeostasis (Sonic hedgehog (SHH), Wnt, TGFbeta, cAMP; [1]). Primary cilium dysfunctions result from an expanding group of rare genetic diseases referred to as ciliopathies, in which nearly all organs can be affected. Associations of these clinical features define about 20 rare syndromes. However, collectively ciliopathies affect up to 1 in 2000 people. Over the past two decades, more than 90 genes have been reported as mutated in ciliopathy patients. Most proteins encoded by these genes play key roles in the biogenesis or function of cilia, in which they define different functional subdomains. Genetic analyses of ciliopathies revealed a vast clinical variability and a broad genetic heterogeneity as: 1) mutations of the same disease-causing gene can result in distinct clinical entities and, conversely, 2) mutations in several independent genes can lead to similar clinical features, implying both phenotypic and genetic overlaps. The extent and severity of organ involvement may be correlated in part with the nature or location of the mutational event, the cell/tissue specific expression and effect of the mutated protein on cilia dysfunction.
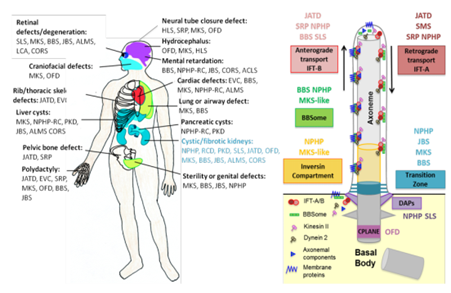
Ciliary protein networks have been correlated to ciliopathy phenotypes. Among these ciliary functional networks, the transition zone plays a role of a gatekeeper; the inversin compartment is involved in ciliary signaling (Wnt, Calcium); IFT proteins are involved in intra-flagellar transport; centriolar proteins (DAPs and CPLANE) are required for ciliogenesis; and BBSome is involved in ciliary protein trafficking with IFT. ACLS: Acrocallosal syndrome; ALMS: Alström syndrome; BBS: BardetBiedl syndrome; CORS: cerebello-oculo-renal syndrome; EVC: Ellis van Creveld syndrome; HLS: Hydrolethalus syndrome; JATD:Jeune Asphyxiating Thoracic Dysplasia; JBS: Joubert syndrome; LCA: Leber's congenital amaurosis; MKS: Meckel syndrome; MZSD: Mainzer-Saldino syndrome; NPHP: Nephronophthisis; OFD: Oral-facial-digital syndrome; SLS: Senior–Løken syndrome; SRP: Short-rib polydactyly syndrome; RCD: Renal cystic dysplasia.CPLANE: Ciliogenesis and planar polarity effector; DAPs: Distal appendage proteins. IFT: Intraflagellar transport.
Renal involvement is one of the most frequent manifestations in ciliopathies, and it leads to excessive morbidity and mortality. This includes renal cystic dysplasia (RCD), a kidney developmental defect, and nephronophthisis (NPHP), a chronic tubulointerstitial nephritis, both disorders representing frequent causes of end-stage renal disease (ESRD) during childhood to early adulthood [2]. While chronic kidney disease (CKD) can be an isolated clinical feature, it also occurs as part of most ciliopathy syndromes (15-30% of BBS, JATD, JBTS, retinal dystrophy patients), also inevitably leading to ESRD despite with a broad timeframe variability. This makes ESDR a terminal endpoint of either isolated or syndromic ciliopathies, with, hitherto, no available curative treatment of chronic kidney disease whatsoever. The only bearable option is renal transplantation. As the average life-span of a functioning kidney transplant is about 10-15 years, it is urgent to identify therapeutic solutions that slow down progression of CKD in ciliopathies, and delay or avoid dialysis or transplantation.
Today, the diagnosis of ciliopathies is first based on primary clinical manifestations, and then confirmed by gene mutation identification. However, even in patients with identified causative mutations, it is impossible to predict the severity of the disease, the risk of appearance (if not present at diagnosis), and/or the rate of progression of CKD. As an example, patients with severe early onset retinal dystrophy have a high risk of developing CKD when carrying NPHP5 mutations, but these mutations are not predictive of the disease progression rate (Otto et al., 2005). Thus, another crucial issue in the field of ciliopathies is to be able to perform early detection of at-risk patients prior to development of CKD as well as to predict disease progression rate.
Identification of more accurate genotypic criteria (including modifiers) as well as better characterization of the phenotypic spectrum associated with mutations in ciliopathy genes are thus dramatically required to allow/improve diagnosis, prognosis, personalized follow-up, and treatment, with an early identification of at-risk patients.
AI has been proven to be a promising approach to decipher such medical complex systems, in particular for the development of predictive models and decision making, as shown by [3; 4; 5].
However, machine-learning techniques obtain optimal results in the presence of big data, which will not be the case in C’IL-LICO project. This is why a more integrative approach will be developed: learning from a limited number of heterogeneous cases, with complex phenotypes and complex underlying biological mechanisms. This approach will be further applied to a target population that will be concurrently readjusted during the project, as data accumulate and we get new results. AI will provide multi-level computational models to help researchers and clinicians in their understanding of ciliopathies, thus allowing early detection of patients and better access to personalized care.
[1] Goetz SC et al. Nat Rev Genet. 2010 May;11(5):331-44.
[2] Salomon R, Saunier S et al. Pediatric nephrology. 2009;24(12):2333-44.
[3] Kourou K et al. Comput Struct Biotechnol J. 2014 Nov 15;13:8-17.
[4] Esteva A et al. Nature. 2017 Feb 2;542(7639):115-118.
[5] Xu G et al. Gene. 2017 Mar 10;604:33-40.
 Français
Français
 English
English

